
BIOSTATISTICS-Normal Curve, Test of significance, Standard error
smaller
BIOSTATISTICS-Normal Curve, Test of significance, Standard error Read More »

Renal Trauma Q. 1 Which of the following is true about renal trauma‑ A Urgent IVP is indicated B Exploration of the kidney to be done in all cases C Lumbar approach to kidney is preferred D Renal artery aneurysm is common Q. 1 Which of the following is true about renal trauma‑ A Urgent

Odontogenic keratocyst Q. 1 Which of the following jaw cyst is pre-malignant? A Nasopalatine cyst B Radicular cyst C Odontogenic keratocyst D Dentigerous cyst Q. 1 Which of the following jaw cyst is pre-malignant? A Nasopalatine cyst B Radicular cyst C Odontogenic keratocyst D Dentigerous cyst Ans. C Explanation: Ans. c. Odontogenic keratocyst Odontogenic keratocysts
Odontogenic Keratocyst Read More »

Pediatric burn injury Q. 1 A 3 year old child suffers from burn injury with the following body parts involved: face including scalp, both buttocks and circumferentially around both thighs. How much is TBSA involved? A 0.25 B 0.26 C 0.35 D 0.45 Q. 1 A 3 year old child suffers from burn injury with
Pediatric burn injury Read More »

Atherosclerosis Q. 1 All will predispose to atherosclerosis except: A Homocystinemia B Fibrinogen C Calcium D Lipoprotein A Q. 1 All will predispose to atherosclerosis except: A Homocystinemia B Fibrinogen C Calcium D Lipoprotein A Ans. C Explanation: Q. 2 Atherosclerosis in the coronary circulation most commonly affects – A Left anterior descending artery B

Mitochondrial DNA Q. 1 Mitochondrial DNA is: A Closed circular B Nicked circular C Linear D Open circular Q. 1 Mitochondrial DNA is: A Closed circular B Nicked circular C Linear D Open circular Ans. A Explanation: Mitochondrial DNA is a closed circular double helix molecule which is transmitted maternally and is found within cells
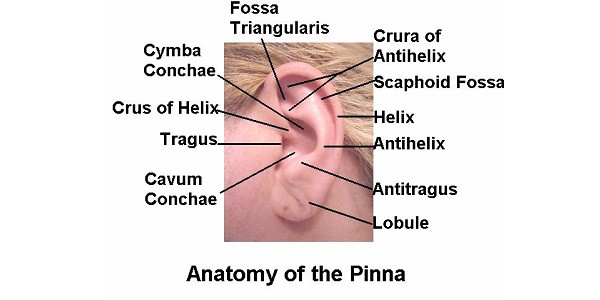
Pinna / Auricle Q. 1 Ear pinna develops from ____________ A Ectoderm B Endoderm C Mesoderm D All Q. 1 Ear pinna develops from ____________ A Ectoderm B Endoderm C Mesoderm D All Ans. A Explanation: Ans:A.)Ectoderm First branchial cleft is the precursor of external auditory canal. Around the sixth week of embryonic life, a
External Ear- Pinna / Auricle Read More »

(VON RECKLINGHAUSEN’S DISNEUROFIBROMATOSISEASE) Q. 1 All are seen in neurofibromatosis EXCEPT? A Meningioma B Lisch nodule C Axillary freckling D Shagreen patch Q. 1 All are seen in neurofibromatosis EXCEPT? A Meningioma B Lisch nodule C Axillary freckling D Shagreen patch Ans. D Explanation: Shagreen patch REF: Harrison’s 17th ed chapter 374 “Shagreen patch is seen
Von Recklinghausen’s Disneurofibromatosisease Read More »
