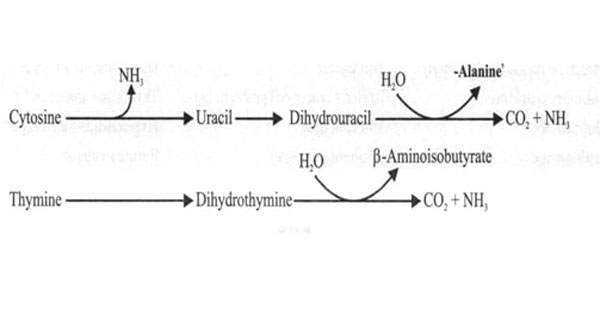
Degradation of Purine Nucleotides
Degradation of Purine Nucleotides Q. 1 Uric acid is formed in humans in A Liver B Kidney C GIT mucosa D Joints Q. 1 Uric acid is formed in humans in A Liver B Kidney C GIT mucosa D Joints Ans. B Explanation: GIT mucosa Most of the dietary purines are converted to uric acid […]
Degradation of Purine Nucleotides Read More »