HEMOGLOBINOPATHIES
HEMOGLOBINOPATHIES
- It is the family of genetic disorders caused by production of a structurally abnormal hemoglobin molecule, synthesis of insufficient quantities of normal hemoglobin, or, rarely, both.
- variants leading to hemoglobinopathies may be either alpha chain variants or beta chain variants.
- Abnormalities in the primary sequence of globin chains lead to hemoglobinopathies,e.g. HbS (qualitative hemoglobinopathy)
- Abnormalities in the rate of synthesis would result in thalassemias (quantitative hemoglobinopathy).
Sickle cell anemia (Hb S), hemoglobin C disease (Hb C), hemoglobin SC disease (Hb S + Hb C), and the tha-lassemia syndromes are representative hemoglobinopathies that can have severe clinical consequences.
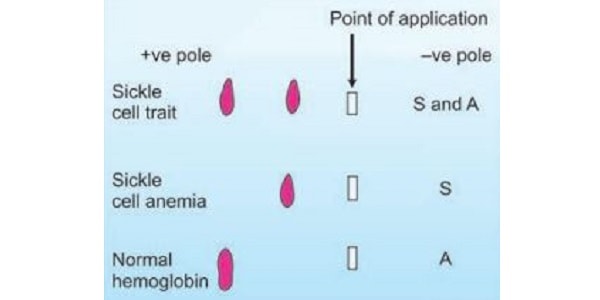
1.Sickle syndromes
A. Sickle-cell trait (AS):
- heterozygous (AS) condition, 50% of Hb in the RBC is normal. Therefore the sickle cell trait as such does not produce clinical symptoms.
- hypoxia may cause manifestation of the disease. Chronic lung disorders may also produce hypoxia-induced sickling in HbS. trait.
- HbS affords protection against Plasmodium falciparum infection
In sickle cell trait, both the bands of HbA and HbS can be noticed in electrophoresis.

B. Sickle-cell disease with SS, SC, SD, SO varieties and S beta thalassemia.
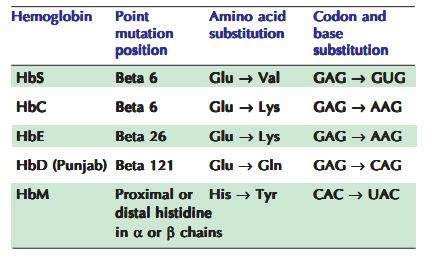
- glutamic acid in the 6th positionof beta chain of HbA is changed to valine in HbS causing a distortion of cell into sickle shape.
- HbA and HbF will prevent sickling, because they do not co-polymerize with HbS. sickling occurs under deoxygenated state.
Congenital Heinz body anemia:
- Unstable Hb variants have an increased tendency to denature which lead to increased hemolysis.
- Heinz bodies are stained purple with cresyl violet. Their occurrence in RBC indicates that the cells have been subjected to oxidative stress.
- Heinz bodies alter the surface of the red cells, creating indentations. As a result, they have a tendency of getting trapped in the spleen.(life-span of RBC reduced).
3. Hemoglobins with abnormal oxygen affinity
A. High affinity—Polycythemia (familial): Hb Chesapeake. Hb binds oxygen, but has difficulty in unloading. The ODC is shifted to the left, with a diminished Bohr effect. So, tissues suffer from hypoxia.
B. Low affinity—Cyanosis (familial): HbM–>occurs in the proximal or distal histidine residues of alpha or beta chains.
–> Alpha 58 His →Tyr (Hb M Boston)
–> Beta 92 His →Tyr (Hb M Hyde Park)
4. Structural variations leading to thalassemia
A. Alpha thalassemia—Hb Constant spring, delta beta thalassemia, Hb Lepore
B. Beta thalassemia: Hb Quong sze
5. Non-symptomatic hemoglobin variants:- HbP, Q, N, J, etc.
