
CONGENITAL / DEVELOPMENTAL GLAUCOMA
smaller
CONGENITAL / DEVELOPMENTAL GLAUCOMA Read More »

Renal Trauma RENAL INJURY Renal trauma is due to- a) Minor injuries- blunt trauma (RTA, falls, assaults & sporting injuries) b) Major injuries – penetrating trauma (knife or gunshot wounds) Blunt trauma is much more common than penetrating trauma. Renal injuries are classified as follows- 1. Grade I- Contusion or non-enlarging subcapsular perirenal haematoma, and

Odontogenic Keratocyst ODONTOGENIC KERATOCYST OKC arises from residual strands of epithelium from dental lamina. Forms a cyst in the jaw in tooth bearing area. Lined by keratinized squamous epithelium. It is locally aggressive and high rate of recurrence OKC –> ameloblastoma –> malignant lesion (SCC) Clinical features- Patient between 10- 40 years are affected.
Odontogenic Keratocyst Read More »

Pediatric burn injury INTRODUCTION: Burns and scalds account for 6% of peadiatric injuries. The majority involve pre-school children,burns being most common between 1-2 yrs,flame burns bet 5-18 yrs. Children have nearly 3 times BSA:BM ratio of adults. Consequently greater fluid requirements and more evaporative water loss than adults. Children Burn that may appear partial thickness
Pediatric burn injury Read More »

Atherosclerosis ATHEROSCLEROSIS Atherosclerosis is an thickening & hardening muscular arteries which is characterised by soft gramous lipid cores (atheromatous plaque). It is the commonest & most important arterial disease. Most commonly affected are aorta, coronaries & cerebral arterial system. Most commonly coronoary circulation affected is – Left anterior descending artery Etiology– Pathogenesis- Most widely accepted

Mitochondrial DNA Mitochondrial DNA (mtDNA) It is a separate genome located in the cytoplasm of nearly all eukaryotic cells Is closede circular, double-stranded, and composed of heavy (H) and light (L) chains or strands. Contains 16,569 bp. Encodes 13 protein subunits of the respiratory chain (of a total of about 67) – Seven subunits of NADH
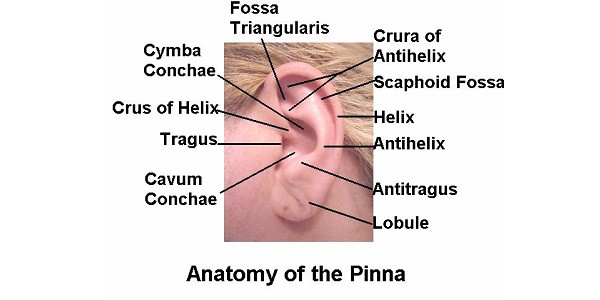
EXTERNAL EAR- Pinna / Auricle AURICLE OR PINNA The entire pinna (except its lobule and outer part of external acoustic canal) is made up of a framework of a single piece of yellow elastic cartilage There is no cartilage between the tragus and crus of the helix – incisura terminalis An incision made in this
External Ear- Pinna / Auricle Read More »

(VON RECKLINGHAUSEN’S DISNEUROFIBROMATOSISEASE) NEUROFIBROMATOSIS (VON RECKLINGHAUSEN’S DISEASE) Neurofibromatosis type 1 (NF1) and type 2 (NF2) are neurocutaneous disorders inherited as autosomal dominant genetic syndromes.. Neurofibromatosis type 1 The clinical criteria used to diagnose NF1 are as follows, in the absence of alternative diagnoses: Six or more café-au-lait spots or hyperpigmented macules =5 mm in diameter
Von Recklinghausen’s Disneurofibromatosisease Read More »

Bioterrorism Agent DEFINIITION: Bioterrorism is terrorism involving the intentional release or dissemination of biological agents. These agents are bacteria, viruses, fungi, or toxins, and may be in a naturally occurring or a human-modified form, in much the same way in biological warfare. Sentinel laboratories are mainly involved in Control of Bioterrorism Category A Definition The U.S. public health system and primary healthcare providers must
Bioterrorism Agent Read More »

Salvage pathway of purine nucleotide synthesis Q. 1 The purines salvage pathway is for: A Hypoxanthine and Xanthine B Hypoxanthine andAdenine C Adenine and Guanine D Xanthine and Guanine Q. 1 The purines salvage pathway is for: A Hypoxanthine and Xanthine B Hypoxanthine andAdenine C Adenine and Guanine D Xanthine and Guanine Ans. B Explanation:Hypoxanthine
Salvage Pathway Of Purine Nucleotides Read More »