
Renal Stones/ Nephrolithiasis
smaller
Renal Stones/ Nephrolithiasis Read More »

Degradation of Purine Nucleotides Q. 1 Uric acid is formed in humans in A Liver B Kidney C GIT mucosa D Joints Q. 1 Uric acid is formed in humans in A Liver B Kidney C GIT mucosa D Joints Ans. B Explanation: GIT mucosa Most of the dietary purines are converted to uric acid
Degradation of Purine Nucleotides Read More »
Degradation of Purine Nucleotides CATABOLISM OF PURINE NUCLEOTIDES Human catabolises purine to uric acid. In higher primates, Allantoin by enzyme uricase is the end product. Adenine nucleotides catabolism- liver, heart muscle, Skeletal muscle, GIT mucosa. Guanine nucleotides catabolism- liver, spleen, kidney, pancreas, GIT mucosa. First metabolic product of purines is Xanthine. Exam Important Human catabolise
Degradation of Purine Nucleotides Read More »
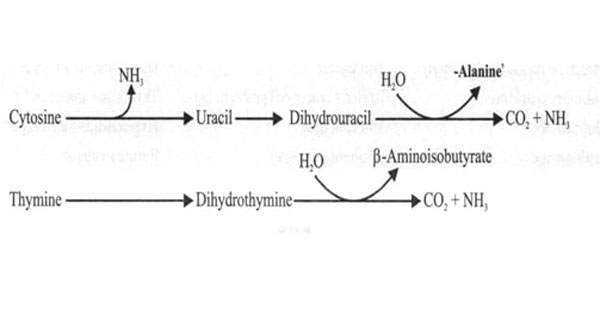
Degradation of Pyrimidine Nucleotides Q. 1 What is the end product of catabolism of pyrimidine? A NH3 B CO2 & H2O C Both D None Q. 1 What is the end product of catabolism of pyrimidine? A NH3 B CO2 & H2O C Both D None Ans. B Explanation: The end products of pyrimidine catabolism is CO2 and
Degradation of Pyrimidine Nucleotides Read More »
Degradation of Pyrimidine Nucleotides CATABOLISM OF PYRIMIDINE NUCLEOTIDES Cytosine and Uracil to Beta Alanine — CO2, NH3 Thymine and β- aminoisobutyrate — CO2, NH3 The end products are highly water soluble. Exam Important The end products of pyrimidine catabolism are highly water soluble. E.g. CO2, NH3, β- aminoisobutyrate, beta alanine. Pseudouridine is excreted unchanged as
Degradation of Pyrimidine Nucleotides Read More »
Steps Of Protein Synthesis (Translation) Q. 1 Which of the following is required for certain types of eukaryotic protein synthesis but not for prokaryotic protein synthesis? A Ribosomal RNA B Messenger RNA C Signal recognition particle D Peptidyl transferase Q. 1 Which of the following is required for certain types of eukaryotic protein synthesis
Steps Of Protein Synthesis (Translation) Read More »
STEPS OF PROTEIN SYNTHESIS (Translation) TRANSLATION Translation is the process in which the genetic information stored in DNA is passed on to mRNA where it is translated into proteins. Translation occurs in ribosomes. mRNA is translated from its 5’ end to its 3’- end (51 à 31 ) 4 letter language information from nucleic acids
Steps Of Protein Synthesis (Translation) Read More »
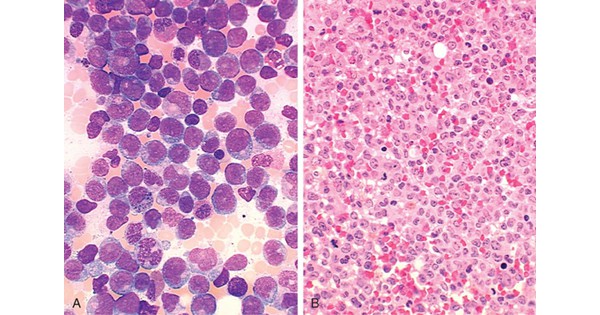
Acute myeloid leukemia (AML) Q. 1 Non-specific esterase is positive in all the categories of Acute Myeloid Leukemia, EXCEPT: A M3 B M4 C M5 D M6 Q. 1 Non-specific esterase is positive in all the categories of Acute Myeloid Leukemia, EXCEPT: A M3 B M4 C M5 D M6 Ans. A Explanation: Non specific
Acute myeloid leukemia (AML) Read More »
Acute myeloid leukemia (AML) ACUTE MYELOID LEUKEMIA (AML) AML is a heterogenous disease characterised by infiltration of malignant myeloid cells into blood, bone marrow. AML is due to inhibition of maturation of myeloid stem cells due to mutations. Seen in mainly in adults (50 years). Chromosomal mutations in AML are translocation t (8: 21) &
Acute myeloid leukemia (AML) Read More »

Sjogren syndrome Q. 1 Biopsy of the parotid gland in a patient with Sjogren’s syndrome shows – A Neutrophils B Lymphocytes C Eosinophi Is D Basophils Q. 1 Biopsy of the parotid gland in a patient with Sjogren’s syndrome shows – A Neutrophils B Lymphocytes C Eosinophi Is D Basophils Ans. B Explanation: Ans. is